The Group
About Us
People
Contacts
Industries
Life Science
Beverage
Cosmetics
Food
Rigid Containers
DIAMIND Solutions
Line – Inspection
Line – Track&Trace
Factory
Warehouse
Enterprise
Supply Chain
Investors
Investor Relations
Governance
Sustainability
Sustainability
Sustainability Strategy
Sustainability Report
Sustainability Initiatives / Projects
Social commitment and education
Contacts
Media
Press Release
Publications
Blog
News
Events
it
en
en
change language
Italiano
English
Blog
Home
|
Blog
14 December 2023
#Supply Chain
REVOLUTIONIZING THE CIRCULAR ECONOMY: UNLEASHING THE POWER OF THE DIGITAL PRODUCT PASSPORT (DPP) THROUGH ADVANCED DIGITALISATION
READ ALL
14 December 2023
#Luxury
ANTARES VISION GROUP AND STIMA: ENSURING SECURITY, AUTHENTICITY, AND VALUE OF LUXURY GOODS IN REAL-TIME
READ ALL
17 November 2023
#Cosmetics
#Beverage
#Food
#Lifescience
DESIRE FOR AWARENESS: FROM THE BARCODE TO THE NEW GS1 QR-CODE IN 2027
READ ALL
15 November 2023
#Food
CIBUS TEC INNOVATION AWARD 2023: ANTARES VISION GROUP RECOGNIZED FOR MICROWAVE TECHNOLOGY
READ ALL
15 November 2023
#Lifescience
THE ANTARES VISION GROUP AUTOMATED MEDICATION CART SELECTED FOR THE ADI DESIGN INDEX IN THE “DESIGN FOR WORK” CATEGORY
READ ALL
15 November 2023
#Finance
THE BOARD OF DIRECTORS APPROVES CONSOLIDATED REVENUES AS AT 30 SEPTEMBER 2023
READ ALL
18 October 2023
#Supply Chain
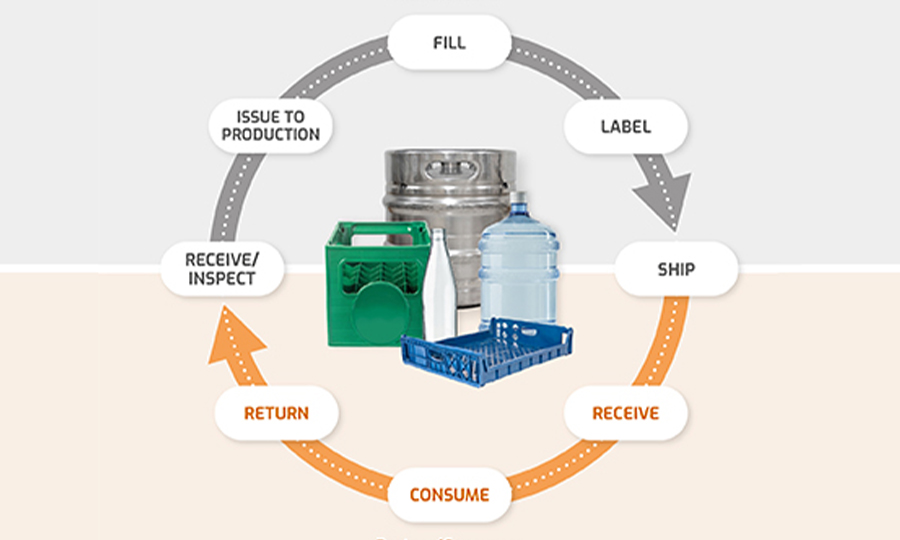
ANTARES VISION GROUP, THANKS TO ACSIS TECHNOLOGY, RELEASES NEW, NEXT-GEN VERSION OF RETURNABLE ASSET MANAGEMENT SOFTWARE
READ ALL
17 October 2023
#Corporate
THE ANTI-COUNTERFEITING TECHNOLOGY CAN BRING TOGETHER GOVERNMENTS, ENTERPRISES, AND CONSUMERS TO FIGHT COUNTERFEIT PRODUCTS
READ ALL
16 October 2023
#Corporate
EMIDIO ZORZELLA, ANTARES VISION GROUP’S CHAIRMAN, JOINS THE HEALTHCARE PANEL FOR THE SUB-SAHARAN AFRICA BUSINESS & INVESTMENT SUMMIT AT THE UNITED NATIONS
READ ALL
15 September 2023
#Lifescience
ANTARES VISION GROUP IN GARTNER HYPE CYCLE 2023 FOR LIFE SCIENCE MANUFACTURING, QUALITY AND SUPPLY CHAIN
READ ALL
14 September 2023
#Finance
THE BOARD OF DIRECTORS APPROVES THE CONSOLIDATED RESULTS AS AT 30 JUNE 2023
READ ALL
13 July 2023
#Supply Chain
Returnable asset and returnable packaging management: Antares Vision Group’s solution for efficiency, quality and safety
READ ALL
12 June 2023
#Corporate
ANTARES VISION GROUP AT FORUM FOOD 2023 BY AMBROSETTI CLUB: TRACEABILITY AS A DRIVER FOR CONSCIOUS SUSTAINABILITY
READ ALL
12 May 2023
#Finance
ANTARES VISION GROUP: RESULTS AT 31 MARCH 2023
READ ALL
05 May 2023
INTELLIGENCE AND INNOVATION TOGETHER: “DIAMIND POWERING PRODUCTS AND SUPPLY CHAINS” – THE INTEGRATED ECOSYSTEM OF SOLUTIONS TO SIMPLIFY THE TECHNOLOGY ENVIRONMENT AND HELP BUSINESSES GROW
READ ALL
02 May 2023
#Corporate
ANTARES VISION GROUP ACQUIRES SMART POINT TECHNOLOGIES, A SOFTWARE DEVELOPMENT COMPANY, TO ACCELERATE SAAS BUSINESS- MODEL ACROSS INDUSTRIES
READ ALL
20 April 2023
#Corporate
MAKE INNOVATION AND INTELLIGENCE RUN TOGETHER: DISCOVER THE NEXT-GEN OF PRODUCT AND SUPPLY CHAIN DIGITALIZATION
READ ALL
18 April 2023
#Beverage
LASER SPECTROSCOPY: THE INNOVATIVE APPLICATION FOR NON-INVASIVE DETECTION OF MICRO-ORGANISMS IN PACKAGED LIQUID PRODUCTS
READ ALL
13 April 2023
#Lifescience
ANTARES VISION GROUP, THROUGH RFXCEL, BEGINS PARTNERSHIP WITH RENOWN HEALTH NETWORK FOR DSCSA-COMPLIANT PHARMACEUTICAL TRACKING
READ ALL
10 April 2023
#Food
MICROWAVE TECHNOLOGY: INNOVATIVE INSPECTION TO DETECT PHYSICAL CONTAMINANTS
READ ALL
06 April 2023
#Food
ANTARES VISION GROUP TECHNOLOGY ALLOWS THE TRACEABILITY OF DAIRY SUPPLY CHAIN
READ ALL
1
2
3
k-arrow-blu-right
![Blog [1] - Antares Vision Group](https://antaresvisiongroup.com/wp-content/uploads/2022/11/blog-desk.jpg)
![Blog [2] - Antares Vision Group](https://antaresvisiongroup.com/wp-content/uploads/2022/11/blog-mob.jpg)